

| ųą╬─├¹ĘQ | Ń~▐D▀\Ą░░ū┘|αµ£┐╣¾w |
| äe ├¹ | ATP 7A; ATPase Copper Transporting Alpha Polypeptide; ATPase Cu++ transporting alpha polypeptide (Menkes syndrome); ATPase Cu++ transporting alpha polypeptide; Copper pump 1; Copper transporting ATPase 1; Cu++ transporting P type ATPase; MC 1; MC1; Menkes disease-associated protein; Menkes syndrome; MK; MNK; OHS; ATP7A_HUMAN. |
| 蹊┐ŅIė“ | ├Ōę▀īW ═©Ą└Ą░░ū ▐D▀\Ą░░ū |
| ┐╣¾wüĒį┤ | Rabbit |
| ┐╦┬ĪŅÉą═ | Polyclonal |
| Į╗▓µĘ┤æ¬ | Human, Mouse, Rat, (predicted: Dog, Cow, Horse, Rabbit, ) |
| «aŲĘæ¬ė├ | ELISA=1:500-1000 IHC-P=1:100-500 IHC-F=1:100-500 Flow-Cyt=2ug/Test IF=1:100-500 Ż©╩»Ž×ŪąŲ¼ąĶū÷┐╣įŁą▐Å═Ż® not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. |
| Ęų ūė ┴┐ | 163kDa |
| ╝Ü░¹Č©╬╗ | ╝Ü░¹Ø{ ╝Ü░¹─ż |
| ąį ĀŅ | Liquid |
| ØŌ Č╚ | 1mg/ml |
| ├Ō ę▀ įŁ | KLH conjugated synthetic peptide derived from human ATP7A:242-285/1500 |
| üå ą═ | IgG |
| ╝ā╗»ĘĮĘ© | affinity purified by Protein A |
| ā” ┤µ ę║ | 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. |
| ▒Ż┤µŚl╝■ | Shipped at 4Īµ. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. |
| PubMed | PubMed |
| «aŲĘĮķĮB | Copper-transporting ATPase 1 is an integral membrane protein cycling constitutively between the trans-golgi network and the plasma membrane. It may supply copper to copper-requiring proteins within the secretory pathway, when localized in the trans-golgi network. Under conditions of elevated extracellular copper, it relocalized to the plasma membrane where it functions in the efflux of copper from cells. Defects in ATP7A are the cause of Menkes syndrome; also known as kinky hair disease, an X-linked recessive disorder. Function: May supply copper to copper-requiring proteins within the secretory pathway, when localized in the trans-Golgi network. Under conditions of elevated extracellular copper, it relocalized to the plasma membrane where it functions in the efflux of copper from cells. Subunit: Monomer. Interacts with PDZD11. Subcellular Location: Golgi apparatus. trans-Golgi network membrane; Multi-pass membrane protein. Cell membrane; Multi-pass membrane protein. Note: Cycles constitutively between the trans-Golgi network (TGN) and the plasma membrane. Predominantly found in the TGN and relocalized to the plasma membrane in response to elevated copper levels. Isoform 3: Cytoplasm. cytosol. Isoform 5: Endoplasmic reticulum. Tissue Specificity: Found in most tissues except liver. Isoform 3 is widely expressed including in liver cell lines. Isoform 1 is expressed in fibroblasts, choriocarcinoma, colon carcinoma and neuroblastoma cell lines. Isoform 2 is expressed in fibroblasts, colon carcinoma and neuroblastoma cell lines. DISEASE: Menkes disease (MNKD) [MIM:309400]: An X-linked recessive disorder of copper metabolism characterized by generalized copper deficiency. MNKD results in progressive neurodegeneration and connective-tissue disturbances: focal cerebral and cerebellar degeneration, early growth retardation, peculiar hair, hypopigmentation, cutis laxa, vascular complications and death in early childhood. The clinical features result from the dysfunction of several copper-dependent enzymes. A mild form of the disease has been described, in which cerebellar ataxia and moderate developmental delay predominate. Note=The disease is caused by mutations affecting the gene represented in this entry. Occipital horn syndrome (OHS) [MIM:304150]: An X-linked recessive disorder of copper metabolism. Common features are unusual facial appearance, skeletal abnormalities, chronic diarrhea and genitourinary defects. The skeletal abnormalities include occipital horns, short, broad clavicles, deformed radii, ulnae and humeri, narrowing of the rib cage, undercalcified long bones with thin cortical walls and coxa valga. Note=The disease is caused by mutations affecting the gene represented in this entry. Distal spinal muscular atrophy, X-linked, 3 (DSMAX3) [MIM:300489]: A neuromuscular disorder. Distal spinal muscular atrophy, also known as distal hereditary motor neuronopathy, represents a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. Note=The disease is caused by mutations affecting the gene represented in this entry. Similarity: Belongs to the cation transport ATPase (P-type) (TC 3.A.3) family. Type IB subfamily. Contains 6 HMA domains. SWISS: Q04656 Gene ID: 538 Database links: Entrez Gene: 538 Human Entrez Gene: 11977 Mouse Entrez Gene: 24941 Rat Omim: 300011 Human SwissProt: Q04656 Human SwissProt: Q64430 Mouse SwissProt: P70705 Rat Unigene: 496414 Human Unigene: 254297 Mouse Unigene: 10554 Rat Important Note: This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications. |
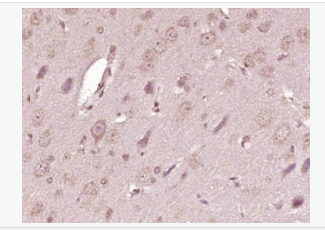
| «aŲĘłDŲ¼ | 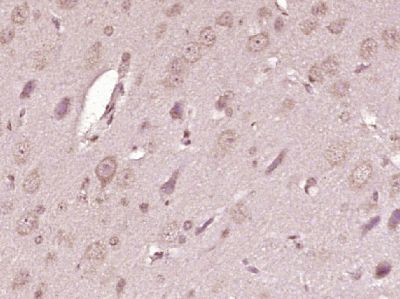 Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (ATP7A) Polyclonal Antibody, Unconjugated (bs-1572R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining. Paraformaldehyde-fixed, paraffin embedded (Rat brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (ATP7A) Polyclonal Antibody, Unconjugated (bs-1572R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.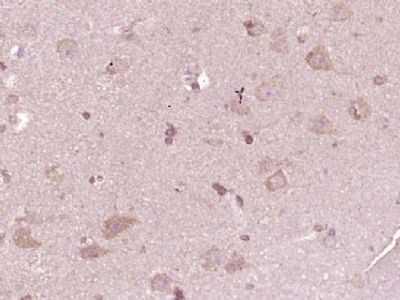 Paraformaldehyde-fixed, paraffin embedded (Human brain glioma); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (ATP7A) Polyclonal Antibody, Unconjugated (bs-1572R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining. Paraformaldehyde-fixed, paraffin embedded (Human brain glioma); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (ATP7A) Polyclonal Antibody, Unconjugated (bs-1572R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.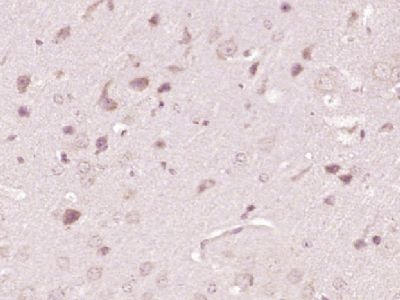 Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (ATP7A) Polyclonal Antibody, Unconjugated (bs-1572R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining. Paraformaldehyde-fixed, paraffin embedded (Mouse brain); Antigen retrieval by boiling in sodium citrate buffer (pH6.0) for 15min; Block endogenous peroxidase by 3% hydrogen peroxide for 20 minutes; Blocking buffer (normal goat serum) at 37°C for 30min; Antibody incubation with (ATP7A) Polyclonal Antibody, Unconjugated (bs-1572R) at 1:400 overnight at 4°C, followed by operating according to SP Kit(Rabbit) (sp-0023) instructionsand DAB staining.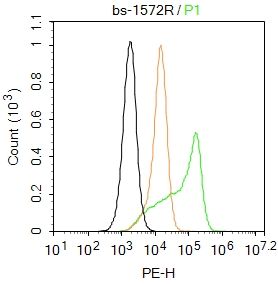 Blank control:U-2OS. Blank control:U-2OS.Primary Antibody (green line): Rabbit Anti-ATP7A antibody (bs-1572R) Dilution: 2μg /10^6 cells; Isotype Control Antibody (orange line): Rabbit IgG . Secondary Antibody : Goat anti-rabbit IgG-PE Dilution: 1μg /test. Protocol The cells were fixed with 4% PFA (10min at room temperature)and then permeabilized with 0.1% PBST for 20 min at room temperature. The cells were then incubated in 5%BSA to block non-specific protein-protein interactions for 30 min at room temperature .Cells stained with Primary Antibody for 30 min at room temperature. The secondary antibody used for 40 min at room temperature. Acquisition of 20,000 events was performed. |
╬ęę¬įāār
*┬ōŽĄĘĮ╩ĮŻ║
(┐╔ęį╩ŪQQĪóMSNĪóļŖūėÓ]ŽõĪóļŖįÆĄ╚Ż¼─·Ą─┬ōŽĄĘĮ╩Į▓╗Ģ■▒╗╣½ķ_)
*ā╚╚▌Ż║





